How Ligands Unlock GPCRs: A Cellular Journey
▼ Summary
– The study describes constructing M2 muscarinic acetylcholine receptor (M2R) biosensors by introducing an amber stop codon for site-specific incorporation of a non-canonical amino acid (TCO*K) via genetic code expansion.
– A two-plasmid system was used in mammalian cells, with one plasmid encoding the receptor mutant and a second encoding an orthogonal tRNA synthetase/tRNA pair to incorporate TCO*K in response to the amber codon.
– The incorporated TCO*K enabled bioorthogonal labeling with fluorescent tetrazine-dye conjugates using rapid click chemistry, allowing the receptors to be visualized and their activity monitored.
– Multiple experimental methods, including live-cell microscopy, ELISA, and BRET-based G-protein activation assays (TRUPATH), were used to characterize the expression, labeling efficiency, and functional responses of the biosensors to various ligands.
– The biosensors were functionally validated by measuring ligand-induced conformational changes linked to G-protein activation and by assessing the effects of perturbations like G-protein overexpression or inhibition with pertussis toxin.
Understanding the intricate dance between G protein-coupled receptors (GPCRs) and their activating ligands is fundamental to cellular communication. This detailed methodology outlines the construction and application of biosensors to visualize and quantify these dynamic interactions in real time within living cells. The approach hinges on genetically encoding a non-canonical amino acid (ncAA) into the receptor, enabling site-specific fluorescent labeling to track conformational changes.
Biosensor construction began with molecular cloning. Most assemblies utilized Gibson assembly with a high-fidelity DNA polymerase, while site-directed mutagenesis introduced amber stop codons at targeted positions within the human M2 muscarinic acetylcholine receptor (M2R) gene. This created receptors with a single, defined site for ncAA incorporation. Various constructs were generated, including versions with cleavable signal peptides, C-terminal eGFP fusions for expression tracking, and N-terminal HA tags for ELISA-based surface expression assays.
Cell culture and transfection protocols were meticulously standardized. HEK293T cells were maintained under standard conditions and seeded onto coated surfaces for imaging experiments. For ncAA incorporation, a two-plasmid system was employed. One plasmid carried the M2R gene with the amber codon, while a second bicistronic plasmid expressed the orthogonal pyrrolysyl-tRNA synthetase (MbPylRS) and its cognate tRNA. This machinery allows the cellular translation system to incorporate the synthetic amino acid trans-cyclooct-4-en-lysine (TCOK) in response to the amber stop codon. Cells were pre-incubated with TCOK before transfection to ensure its availability.
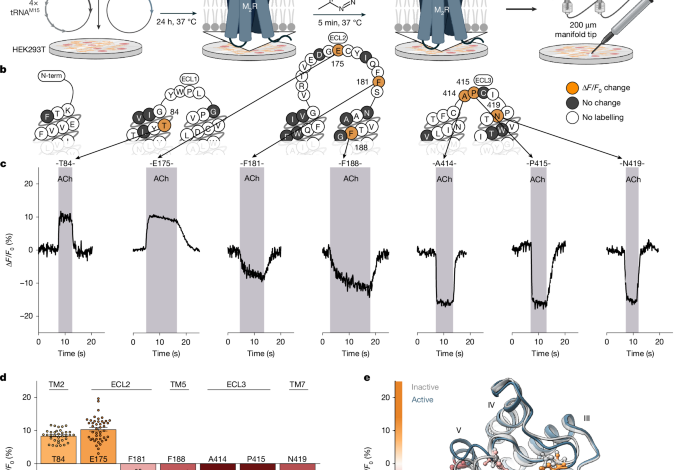
The critical step of bioorthogonal labeling leveraged ultra-rapid click chemistry. The incorporated TCO*K possesses a highly reactive strained alkene, which undergoes a specific and fast inverse electron-demand Diels-Alder reaction with tetrazine-conjugated fluorescent dyes like Cy3 or Cy5. This reaction is exceptionally fast, efficient, and gentle on living cells, enabling precise fluorescent tagging of the receptor at the genetically encoded site. A qualitative screen of numerous M2R mutants identified seven optimal positions for labeling, which were then developed into functional biosensors.
Quantifying labeling efficiency was achieved through temporal brightness analysis. By transfecting cells with the eGFP-tagged biosensor constructs and labeling with Tet-Cy3, researchers could simultaneously monitor total receptor expression (via eGFP) and successfully labeled receptor population (via Cy3). Advanced image analysis calculated the number of emitting molecules per pixel over time, allowing the determination of precise labeling efficiencies for each biosensor, which were crucial for interpreting subsequent functional data.
Functional characterization employed multiple sophisticated assays. Single-cell kinetic microscopy tracked fluorescence changes of the labeled biosensors upon ligand application, revealing real-time conformational shifts. A G i3 FRET activation assay independently measured downstream G-protein activation. Furthermore, a BRET-based TRUPATH assay profiled the specific G-protein subunits activated by the wild-type receptor and each biosensor, with a particular focus on Gα oA activation. Cell-surface ELISAs confirmed that all biosensors were properly trafficked to the plasma membrane.
Data analysis and statistical rigor underpinned all conclusions. Fluorescence changes were normalized to baseline, and concentration-response curves were fitted to determine pharmacological parameters like EC50 and efficacy. Statistical comparisons utilized appropriate t-tests or ANOVA with post-hoc corrections. Molecular modeling helped visualize receptor conformations, and all experiments were repeated independently at least three times to ensure reproducibility. This comprehensive suite of techniques provides a powerful toolkit for dissecting the precise molecular events that occur when a ligand unlocks the functional potential of a GPCR.
(Source: Nature)